Blog post by Dr. Tamara Maiuri
I’ve previously described our hypothesis that huntingtin binds poly ADP ribose and our preliminary evidence that it does so. I now have a few more pieces of data to add to that story.
How does huntingtin bind PAR?
Full length huntingtin, as well as a fragment spanning amino acids 78-426, can bind PAR in vitro in a PAR overlay assay. In an attempt to narrow down which part of the huntingtin protein might bind PAR, I looked at the huntingtin sequence to identify potential PAR-binding motifs (PBMs). Peptides representing four of the most promising PAR-binding motifs (PBMs) were tested for their ability to bind PAR in vitro. PBM3 peptide, which corresponds to amino acids 1782-1804 of the huntingtin sequence (with the PBM at 1790-1798), was the only peptide to bind PAR. The experiment required optimization to maintain peptides on the nitrocellulose membrane. These optimization steps and the results have been deposited to Zenodo.
I was somewhat surprised to find that PBM1 did not bind PAR. That was the only PBM that I could find within the 78-426 fragment, which can bind PAR. It could be that the 78-426 fragment is binding PAR non-specifically, or it could bind PAR through a different motif.
What’s the point of huntingtin PAR-binding?
Current efforts are focused on figuring out the physiological relevance of huntingtin PAR binding. So far, no dice. We asked the following questions:
Is it to stick to chromatin in response to oxidative stress?
Nope.
Truant lab student Carlos Barba tested endogenous huntingtin chromatin recruitment upon enzymatic inhibition of PARP by veliparib. The amount of endogenous huntingtin bound to chromatin after a Triton X-100 wash, which increases upon KBrO3 treatment, was not reduced by inhibition of PARP with veliparib:

Similarly, the amount of transfected huntingtin 1-586 fragment bound to chromatin after a Triton X-100 wash, which increases upon KBrO3 treatment, was not reduced by inhibition of PARP with veliparib (data deposited to Zenodo).
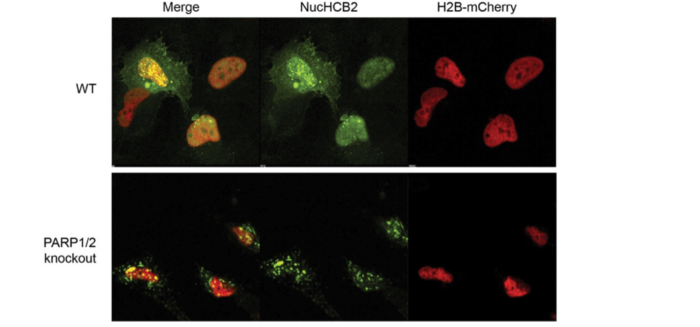
We also found that endogenous huntingtin was still recruited to chromatin in PARP1/PARP2 knockout cells [1] upon oxidative stress. The knockout cells were highly susceptible to stress, so it’s not clear whether huntingtin was bound to chromatin as part of the DNA repair process (which would lead to recovery) or as part of a cell death process. Enzymatic inhibition of PARP3 in the PARP1/PARP2 knockout cells was toxic.
An unexpected result
When I reported to Ray my lack of success in determining why the heck huntingtin binds PAR, he suggested I try looking at the dynamics of huntingtin chromatin retention using Fluorescence Recovery After Photobleaching (FRAP) of the YFP-tagged intrabody that recognizes endogenous huntingtin [2]. I found that KBrO3 slows huntingtin mobility, as would be expected upon huntingtin chromatin binding. If huntingtin uses PAR binding as a mechanism of chromatin retention, then inhibition of PAR formation should restore huntingtin mobility. In contrast, veliparib significantly reduced huntingtin mobility beyond the degree caused by KBrO3 alone. PARP inhibitors are known to “trap” PARP at chromatin, so it’s possible that huntingtin is also being trapped. I will repeat these experiments using PARP knock down strategies to try and clarify what’s happening, and deposit everything to Zenodo.

Is it to get to huntingtin stress bodies (HSBs)?
Nope.
HSBs are spots in the cytoplasm where huntingtin accumulates upon cell stress [3]. There are many types of stress bodies in cells, and some of them use PAR in their formation [4]. I tested whether HSBs could still form in the presence of a PARP inhibitor and found it had no effect on HSB formation.
Is it to interact with PARylated proteins?
The plot thickens…
We set out on this PAR journey because we found that a large proportion of the huntingtin interacting proteins we identified are also part of PARylated protein databases. To make sure that huntingtin does indeed physically interact with PARylated proteins, I performed co-immunoprecipitation experiments. Not surprisingly, huntingtin pulls down many PARylated proteins (experiments deposited to Zenodo). One of those proteins is the PARP enzyme itself, although the conditions promoting huntingtin-PARP interaction were variable (see Zenodo entry). What came as a surprise was that inhibition of PARP with veliparib actually increased the interaction between huntingtin and PARylated proteins, including PARP. It’s possible that this is related to the slowed huntingtin mobility observed in FRAP experiments upon veliparib treatment.
Is it to get into or out of nuclear speckles?
Not sure yet.
Nuclear speckles, or SC35 domains, are sub-nuclear regions of low DNA and high protein where huntingtin lives [5]. We have found PAR at these sites [2], where it is thought to nucleate liquid-liquid phase transition of proteins into this “membraneless organelle” [6]. I am currently conducting experiments to find out whether huntingtin binds PAR to localize to these sites.
Any other ideas?
If anyone has other suggestions for figuring out why the heck huntingtin binds PAR, I am all ears!
- Hanzlikova H, Gittens W, Krejcikova K, Zeng Z, Caldecott KW. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 2017;45: 2546–2557.
- Maiuri T, Mocle AJ, Hung CL, Xia J, van Roon-Mom WMC, Truant R. Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet. 2017;26: 395–406.
- Nath S, Munsie LN, Truant R. A huntingtin-mediated fast stress response halting endosomal trafficking is defective in Huntington’s disease. Hum Mol Genet. 2015;24: 450–462.
- Leung AKL, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol Cell. 2011;42: 489–499.
- Hung CL-K, Maiuri T, Bowie LE, Gotesman R, Son S, Falcone M, et al. A Patient-Derived Cellular Model for Huntington’s Disease Reveals Phenotypes at Clinically Relevant CAG Lengths. Mol Biol Cell. 2018; mbcE18090590.
- Leung AKL. Poly(ADP-ribose): an organizer of cellular architecture. J Cell Biol. 2014;205: 613–619.